پیراسته فر
علمی،تحقیقی و تحلیلیپیراسته فر
علمی،تحقیقی و تحلیلیرابطه پروتئین و پیر شدن سلولها
«تراکم پروتئین» باعث پیری سلولها میشود واین ۲ ارتباط تنگاتنگی با هم دارند.
درصورمختل شدن سیستم ثبات و پایداری پروتئینها «پیری زودرس» اتفاق می افتد.
اگر مسیرهای کنترل کیفیت پروتئین از نظر ژنتیکی افزایش یابند،برفرآیندپیری کاسته خواهدشد.
البته تراکم پروتئین باعث پیری سلولها میشود واین دو ارتباط تنگاتنگی با هم دارند.
رابطه پروتئین و پیر شدن سلولها
بررسی از رابطه بین تراکم پروتئین و پیری.

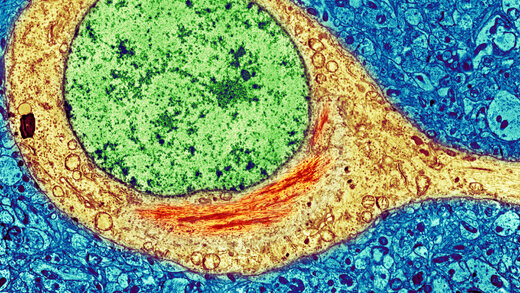
محققان استنفورد، پروتئینهای موجود در انواع «ماهی کیلیفیش» که به طور غیرعادی سریع پیر میشوند را با دقت بیشتری بررسی کردند.
این ماهیها در ژن «آنزیم تلومراز» خود دارای یک جهش هستند که طول «کروموزوم»های تقسیم شده را حفظ میکند. حیواناتی که دارای «جهش تلومراز» هستند معمولا به سرعت پیر میشوند.
«جاروس» گفت که او و همکارانش انتظار داشتند که در روده و سایر بافتهایی که به سرعت رشد میکنند و جایگزین میشوند، تراکم کمتری وجود داشته است:
تقسیم سلولی اضافی به بافتهایی که به سرعت در حال رشد هستند فرصت بیشتری برای پاکسازی تودهها و بازسازی خودشان میدهد.
![]()
اما عکس این مطلب اتفاق افتاد: بافتهایی که به سرعت رشد می کردند، پروتئینهای متراکمتری داشتند و نسبت به بافتهایی که آهسته رشد می کردند، سریعتر پیر میشدند.
بنابراین دوباره مشکلات مربوط به کنترل سلول بر کیفیت پروتئینهایش ممکن است توضیح این مطلب باشد.
اگر سلولها کنترل خود بر فرآیندهایی که کیفیت پروتئینهایشان را حفظ میکنند از دست بدهند، با هر تقسیم سلولی آسیب بیشتری از تراکمها ایجاد خواهد شد.
علت پیری زودرس
بافتهایی که به سرعت رشد میکنند، چون شانسشان برای متراکم کردن آسیب بیشتر میشود، سریعتر پیر میشوند.
علت اینکه چرا گاهی پروتئینها تجمع پیدا میکنند، پیچیده است. در کمال تعجب، بخشی از پاسخ این سوال به طور اساسی به یک مکانیسم ضروری به نام تراکم مرتبط است که سلولها برای کنترل پروتئینهای خود از آنها استفاده میکنند.
این که دقیقا چه چیزی باعث جذب پروتئینها و ایجاد تودهها میشود و این تودهها چقدر برای سلولها مشکل ایجاد میکنند، هنوز یک بحث بزرگ و خارقالعاده در این زمینه است.
بیماری هانگتینتون
نمونه بسیار خوب از اثر محافظتی که از این مطالعات بدست آمده، «پروتئین هانگتینتین» است که برای رشد سالم سیستم عصبی ضروری است، اما در افراد مبتلا به «بیماری هانگتینتون» یک جهش باعث شده تا پروتئین هانگتینتین به طور غیرطبیعی طولانی و بلند شود. سپس این پروتئین بلند به بخشهای کوچک و سمی تقسیم میشوند که به سیستم عصبی آسیب میزند.

تجمع پروتئین هانتینگتین
![]()
در سال ۲۰۰۴، «استیو فینکبینر» محققی که در زمینه پیری در موسسه گلادستون و دانشگاه کالیفرنیا، سن فرانسیسکو بر روی «تجمع پروتئین هانتینگتین» که در سلولهای عصبی کشت شده بود مطالعه ای را انجام داد.

![]()
تیم او در این تحقیق نشان دادند که اگرچه تمام نورونهایی که دارای پروتئینهای غیرطبیعی هانتینگتین بودند، با گذشت زمان مُردند، اما نورونهایی که در آنها تجمع هانتینگتین وجود داشت توانستند نسبت به نورونهایی که این پروتئین را نداشتند، زمان بیشتری را زنده بمانند.
توضیح مدیریت سایت-پیراسته فر: تجمع پروتئین هانتینگتین یک بیماری اتوزومال غالب و پیشرونده نورودژنراتیو است.

![]()
«بیماری هانتینگتون» (HD) یک بیماری اتوزومال غالب و پیشرونده نورودژنراتیو است که در اثر گسترش تکرار سه گانه CAG (> 35) در ژن هانتینگتین ایجاد می شود. این به یک تکرار پلی گلوتامین (polyQ) بلافاصله پس از 17 اسید آمینه اول (N17) در پروتئین هانتینگتین (HTT) تبدیل می شود.
فسفوریلاسیون(Phosphorylation) دامنه N ترمینال پروتئین هانتینگتین (HTT) به عنوان یک تنظیم کننده مهم در محلی سازی، ساختار، تجمع، ترخیص کالا از گمرک و سمیت آن ظاهر شده است. با این حال، اعتبار سنجی اثر فسفوریلاسیون واقعی در داخل بدن و ارزیابی پتانسیل درمانی هدف قرار دادن فسفوریلاسیون برای درمان بیماری هانتینگتون (HD) نیازمند شناسایی آنزیم هایی است که فسفوریلاسیون HTT را تنظیم می کنند. در اینجا، ما کشف و اعتبار یک کیناز، کیناز 1 متصل به TANK (TBK1) را گزارش میکنیم که به طور موثر قطعات HTT تمام طول و N-ترمینال را در شرایط آزمایشگاهی (در S13/S16)، در سلولها (در S13) و در داخل فسفریله میکند. داخل بدن بیان TBK1 در مدل های HD (سلول ها، نورون های اولیه و Caenorhabditis elegans) فسفوریلاسیون اگزون 1 HTT جهش یافته را افزایش می دهد و تجمع و سمیت سلولی آن را کاهش می دهد. ما نشان میدهیم که اثرات محافظت عصبی با واسطه TBK1 به دلیل مهار وابسته به فسفوریلاسیون تجمع اگزون 1 HTT جهش یافته و افزایش پاکسازی اتوفاژیک HTT جهش یافته است. این یافتهها نشان میدهد که تنظیم مثبت و/یا فعالسازی TBK1 با کاهش همزمان سطوح HTT جهشیافته و مسدود کردن تجمع آن، یک استراتژی مناسب برای درمان HD است.

![]()
فسفوریلاسیون پروتئین هانتینگتین در S13 و S16 نقش حیاتی در تنظیم تجمع، پاکسازی و سمیت آن دارد. TBK1، یک کیناز طبیعی که HTT را در S13 فسفریله می کند، منجر به کاهش تجمعات HTT جهش یافته در سلول ها و C. elegans از طریق مکانیزمی می شود که هم به فسفوریلاسیون HTT S13 و هم اتوفاژی نیاز دارد.
شناسایی یک کیناز جدید، TBK1، که به طور موثر HTT نوع وحشی و جهش یافته را در باقیمانده سرین 13 فسفریله می کند.
بیان بیش از حد TBK1 فسفوریلاسیون S13 را در HTT جهش یافته افزایش می دهد، از تجمع آن جلوگیری می کند و باعث تخریب اتوفاژیک دانه های محلول، اما نه از پیش ساخته شده HTT می شود.
بیان بیش از حد TBK1 در برابر سمیت سلولی HTT جهش یافته در مدل های عصبی و C. elegans HD محافظت می کند.
افزایش فسفوریلاسیون در سرین 13 یا ارتقاء ترخیص کالا از گمرک اتوفاژیک HTT جهش یافته نشان دهنده راهبردهای درمانی مناسب برای درمان HD است.

![]()
پروتئینی است که توسط ژنی به نامBDNF یکی از بارزترین عوامل غیرقابل تنظیم در سندرم رت، یک اختلال عصبی رشدی شدید است. این مطالعه نشان میدهد که فسفوریلاسیون هانتینگتین انتقال آکسونی BDNF را تحریک میکند و بسیاری از ویژگیها را در مدل موش Rett بهبود میبخشد، که یک رویکرد درمانی ممکن را پیشنهاد میکند.
بیماریهای نورودژنراتیو

ادامه مقاله کوانتامگزین: از آن زمان او و دیگران نشان دادهاند که واکنشهای تجمعی حمایتی، در سایر بیماریهای «نورودژنراتیو »نیز اتفاق میافتد. این میتواند علت شکستهای پیدر پی آزمایشهای تجربی آلزایمر با هدف قرار دادن پلاکها را توضیح دهد. او در این باره میگوید: اگر پلاکهای «آمیلوئیدی» که معرف این بیماری هستند مانند پروتئینهای محافظتی به هم متصل شوند، شکستن این پلاکها ممکن است بیشتر از آنکه فایده داشته باشد، مضر باشد.
![]()
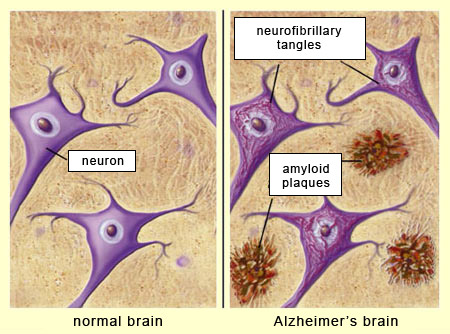
توضیح مدیریت سایت-پیراسته فر:«بیماری های نورودژنراسیون»چیست؟
سلول های مغز ارتباط تنگاتنگی با هم دارند و ارتباط نادرست در یک منطقه می تواند فعالیت های دیگر بخش های مغز را مختل کند واین اختلالات مغزی «بیماری های نورودژنراسیون» نامیده می شوند،بیماری های نورودژنراتیو منجر به مرگ نورون می شوند، این بیماری ها می توانند بر حرکت ، گفتار ، حافظه ، هوش بیمار تأثیر بگذارند،مثل بیماری پارکینسون که یک تخریب عصبی است،در هر بیماری تخریب کننده عصبی ، اگر سلول های عصبی در حال مرگ باشند ،فرآیندکوچک شدن مغز اتفاق می افتد./پایان توضیح.
فینکبینر نوشت: «درک این مفهوم برای انسان دشوار است، زیرا این مطالب شهودی به نظر میرسند، و مفاهیمی که غیر عادی به نظر برسند باید «بد» و «بیماریزا» باشند. اما زیست شناسی علم پیچیدهای است که سرشار از عجایب است. بنابراین بسیار مهم است که مردم در نتیجهگیری فریب نخورند.»
یک چالش عمومی با راه حل های فراوان
نتیجه واضحی که اکنون به آن رسیده ایم این است که تجمع پروتئین پدیدهای مختص به بیماریهای عصبی نیست: این عمل بخشی از هر سلول است که به اندازه کافی عمر کرده و پیر شده است. بسیاری از پروتئینهای طبیعی و مهم مانند DDX5 تمایل به تجمع دارند، و مقابله با این تجمع یک چالش فراگیر است که هر سلول باید به آن رسیدگی کند.
از آنجایی که سلولها برای مدتهای طولانی با این مشکل دست و پنجه نرم کردهاند، پیشگیری از تجمع آنها ممکن است کاری مهم در تکامل توالیهای پروتئین باشد. از آنجایی که تعداد زیادی از پروتئینها آمادگی تجمع کردن را دارند، و جهشها این تمایل را افزایش میدهند، به احتمال بسیار قوی، تعداد زیادی از پروتئینها، به طور طبیعی در مقابل جهش ایستادگی میکنند. (این نتیجه گیری در نتیجه این موضوع است که در حیوانات جوان، بیشتر پروتئینها تمایل کمی به جهش دارند).

![]()
بنابراین پروتئینهایی که تعداد آنها کمتر است ممکن است سریعتر از آنهایی که تعدادشان زیاد است تکامل پیدا کنند و نرخ تکامل آنها باید با تمایلشان به تجمع همخوانی داشته باشد.

![]()
این اثر در مغز «ماهی کیلیفیش» بسیار بارز بود. محققان بر این گمان هستند که این پروتئینها ممکن است کلید نوآوری در اندام بوده باشند. اگر چنین باشد، تغییرات تکامل در مغز که آن را به عضوی مهم در مهرهداران تبدیل کرده است، میتواند این عضو را در برابر بیماریهای تخریبی ناشی از تجمع آسیبپذیرتر کرده باشد.
در واقع این احتمال وجود دارد که هر بافت و اندامی باید به تعادلی متفاوت برای انجام کارهای خود و مدیریت تجمع پروتئین برسد.
هر بافتی نیازها و محدودیتهای عملکردی منحصر بفرد خود را دارد: «سلولهای روده» دائما در حال تغییر هستند؛ سلولهای غُدد درونریز «هورمون» ترشح میکنند؛ «سلولهای مسئول ایمنی بدن» زمانی که مهاجمی را شناسایی میکنند وارد عمل میشوند؛ مغز اطلاعات را پردازش میکند. کارهای مختلف نیاز به پروتئینهای متفاوت دارند و این یعنی استراتژی تکامل یافته برای مقابله با تجمع پروتئین از بافتی به بافت دیگر و از حیوانی به حیوان دیگر متفاوت است.

میتوان گفت از آنجایی که مغز مهره داران در گذشته نه چندان دور بسیار سریعتر و گستردهتر از مثلا عضلات آنها تکامل یافته است، ممکن است دستگاه کنترل کیفیت پروتئین آن هنوز زمان کافی برای تکامل سیستم محافظتی کافی در برابر تجمعهای پروتئینی نسبتا جدید را نداشته باشد.
با این وجود مشکل اساسی «تجمع پروتئین» برای همه موجودات وجود دارد، و این مشکل فقط منحصر به زمانهای بیماری یا استرس شدید نیست و این یک واقعیت است که تجمع پروتئین در سرتاسر بدن عاملی برای پیری در موجودات مختلفی مانند مخمرها، کرمها، مگسها، ماهیها، موشها و انسان است.
توضیح مدیریت سایت-پیراسته فر:منبع اخدشده ،خبرآنلاین می باشد که منبعش (کوانتامگزین)است وبا اصلاحات واضافات./پایان.
اطلاعات تکمیلی:البته باعذرخواهی ازاشکالات درترجمه مقالات اخذشده ازمنابع خارجی.

![]()
اثر پیری در سلول عصبی و مکانیسم های درگیر

![]()
سلول پیری فرآیندهای سلولی مهم در طول پیری تحت تأثیر قرار می گیرند. این منجر به چندین فنوتیپ سلولی، از جمله بار بیش از حد سیستم کنترل کیفیت پروتئین، آسیب DNA، اختلال عملکرد میتوکندری و استرس ER می شود که با هم منجر به آسیب پذیری در برابر مرگ سلولی می شود.
![Proteostasis changes in a senescent cell. The scheme shows the overall changes of the cellular systems maintaining protein homeostasis (proteostasis) and thus, cellular functionality during aging. The main proteolytic systems, responsible for recognition and degradation of un/misfolded or oxidatively damaged proteins are the proteasomal system, involving the ATP-dependent 26S proteasome as well as the ATP-independent 20S proteasome, and autophagy (including both autophagy (MA) as well as the chaperone mediated one (CMA)). Damaged proteins can be directly recognized as substrates by the 20S proteasome, resulting in proteolytic removal from the cell. Furthermore, they can be recognized by chaperones or heat shock proteins that keep their substrates in a soluble state, preventing the formation of aggregates. Another fate might be the formation of aggregates, driven by hydrophobic residues exposure. Such aggregates can be incorporated into an autophagosome and fusing with lysosomes, resulting in proteolytic degradation by lysosomal proteases, or they can be removed from the cell via excretion as exosome. Modified from [115] and according to [122].](https://www.researchgate.net/profile/Jose-Pedro-Castro/publication/311499230/figure/fig6/AS:668652722458628@1536430640394/Proteostasis-changes-in-a-senescent-cell-The-scheme-shows-the-overall-changes-of-the.png)
![]()
پروتئوستاز در یک سلول پیر تغییر می کند. این طرح تغییرات کلی سیستم های سلولی را نشان می دهد که هموستاز پروتئین (پروتئوستاز) و در نتیجه عملکرد سلولی را در طول پیری حفظ می کنند.
سیستمهای پروتئولیتیک اصلی که مسئول شناسایی و تخریب پروتئینهای بدون تاخوردگی یا آسیبدیده اکسیداتیو هستند، سیستم پروتئازومی هستند که شامل پروتئازوم 26S وابسته به ATP و همچنین پروتئازوم 20S مستقل از ATP و اتوفاژی (شامل اتوفاژی (MA) میشود. و همچنین واسطه واسطه (CMA)). پروتئین های آسیب دیده را می توان مستقیماً به عنوان سوبسترا توسط پروتئازوم 20S شناسایی کرد که منجر به حذف پروتئولیتیک از سلول می شود. علاوه بر این، آنها را می توان توسط چپرون ها یا پروتئین های شوک حرارتی که بسترهای آنها را در حالت محلول نگه می دارد و از تشکیل سنگدانه ها جلوگیری می کند، شناسایی کرد. سرنوشت دیگر ممکن است تشکیل سنگدانه ها باشد که توسط قرار گرفتن در معرض پسماندهای آبگریز هدایت می شود. چنین تودههایی را میتوان در اتوفاگوزوم ادغام کرد و با لیزوزومها ترکیب شد و منجر به تخریب پروتئولیتیک توسط پروتئازهای لیزوزومی شد، یا میتوان آنها را از طریق دفع به عنوان اگزوزوم از سلول خارج کرد.
پروتئوستاز (هوموستاز پروتئین)چیست؟
پروتئوستاز (Proteostasis) مجموعههای پروتئینی غیر غشایی هستند که در همه یوکاریوتها و آرکیاها و برخی از باکتریها وجود دارند. عملکرد اصلی پروتئازوم حذف پروتئینهای غیرضروری یا آسیب دیده، توسط پروتئولیز است. پروتئولیز یک واکنش شیمیایی است که پیوندهای پپتیدی را حذف میکند.